










Every complex case is unique, and no calcified lesion is the same. That’s why we focus on providing the therapies, tools, and specialized knowledge that enable you to tailor your approach and optimize long-term, durable outcomes.

Our comprehensive portfolio offers individualized solutions for every step in the treatment of calcified lesions from ACCESS to CLOSE.
Gain access with the right guide wires.
Microcatheters

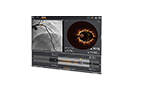
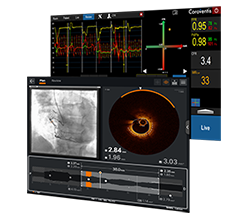
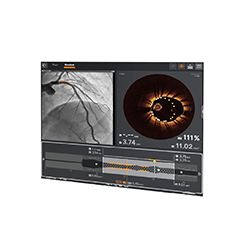
Visualize the best path forward with OCT and full physiology.
Clear obstacles with orbital atherectomy and coronary dilatation catheters.

Request an Abbott Sales Rep
MAT-2414252 v1.0
INDICATIONS FOR USE
This HI-TORQUE™ Guide Wire is intended to facilitate the delivery of catheter-based interventional devices during the following procedures:
This guide wire may also be used with compatible stent devices.
This device is designed and intended for ONE-TIME USE ONLY. Do not resterilize and / or reuse.
CONTRAINDICATIONS
Not intended for use in the cerebral vasculature.
WARNINGS
Not intended for use with atherectomy devices.
Carefully observe the instructions under “Do Not” and “Do” below. Failure to do so may result in vessel trauma, guide wire damage, guide wire tip separation, or stent damage. If resistance is observed at any time, determine the cause under fluoroscopy and take remedial action as needed. Use the most suitable guide wire for the lesion being treated.
Do Not:
Do:
PRECAUTIONS
Guide wires are delicate instruments and should be handled carefully. Prior to use and when possible during the procedure, inspect the guide wire carefully for bends, kinks, or other damage. Do not use damaged guide wires. Using a damaged guide wire may result in vessel damage and / or inaccurate torque response.
Confirm the compatibility of the guide wire diameter with the interventional device before actual use.
Free movement of the guide wire within the interventional device is an important feature of a steerable guide wire system, because it gives the user valuable tactile information. Test the system for any resistance prior to use. Adjust or replace the hemostatic valve with an adjustable valve if it is found to inhibit guide wire movement.
Never attach the torque device to the modified portion of the proximal end of the extendible guide wire; otherwise, guide wire damage may occur, preventing the ability to attach the DOC™ Guide Wire Extension.
HI-TORQUE™ Guide Wires with Hydrophilic Coating: Avoid abrasion of the hydrophilic coating. Do not withdraw or manipulate the hydrophilic-coated wire through a metal cannula or sharp-edged object.
ADVERSE EVENTS
Potential adverse events associated with use of this device may include the following but not limited to perforation, dissection, occlusion, myocardial infarction, embolism and infection.
MAT-2104726 v2.0
INDICATIONS FOR USE
This HI-TORQUETM Guide Wire is intended to facilitate the delivery of catheter-based interventional devices during the following procedures:
• Percutaneous transluminal angioplasty (PTA)
• Percutaneous transluminal coronary angioplasty (PTCA)
This guide wire may also be used with compatible stent devices.
This device is designed and intended for ONE-TIME USE ONLY. Do not resterilize and / or reuse.
CONTRAINDICATIONS
Not intended for use in the cerebral vasculature.
WARNINGS
Not intended for use with atherectomy devices.
Carefully observe the instructions under “Do Not” and “Do” below. Failure to do so may result in vessel trauma, guide wire damage, guide wire tip separation, or stent damage. If resistance is observed at any time, determine the cause under fluoroscopy and take remedial action as needed. Use the most suitable guide wire for the lesion being treated.
Do Not:
Do:
PRECAUTIONS
Guide wires are delicate instruments and should be handled carefully. Prior to use and when possible during the procedure, inspect the guide wire carefully for bends, kinks, or other damage. Do not use damaged guide wires.
Using a damaged guide wire may result in vessel damage and / or inaccurate torque response.
This device should be used only by physicians trained in angiography and percutaneous transluminal coronary angioplasty (PTCA), and / or percutaneous transluminal angioplasty (PTA).
Confirm the compatibility of the guide wire diameter with the interventional device before actual use.
Free movement of the guide wire within the interventional device is an important feature of a steerable guide wire system, because it gives the user valuable tactile information. Test the system for any resistance prior to use. Adjust or replace the hemostatic valve with an adjustable valve if it is found to inhibit guide wire movement.
Never attach the torque device to the modified portion of the proximal end of the extendable guide wire; otherwise, guide wire damage may occur, preventing the ability to attach the DOCTM Guide Wire Extension.
HI-TORQUETM Guide Wires with Hydrophilic Coating: Avoid abrasion of the hydrophilic coating. Do not withdraw or manipulate the hydrophilic-coated wire through a metal cannula or sharp-edged object.
ADVERSE EVENTS
Potential adverse events associated with use ofthis device may include the following but not limited to:
MAT-2104727 v2.0
INDICATIONS FOR USE: Intended to facilitate the delivery of catheter-based interventional devices during percutaneous transluminal coronary angioplasty (PTCA) and percutaneous transluminal angioplasty (PTA). This guide wire may be used with compatible stent devices during therapeutic procedures. The guide wire may be used to reach and cross a target lesion, provide a pathway within the vessel structure, facilitate the substitution of one diagnostic or interventional device for another, and to distinguish the vasculature. This guide wire may also be used to cross or assist in crossing de novo chronic total coronary occlusions (CTO).
CONTRAINDICATIONS: Not intended for use in the cerebral vasculature or with atherectomy devices.
WARNINGS:
This device is designed and intended for ONE-TIME USE ONLY. Do not resterilize and / or reuse.
Carefully observe the instructions under “Do Not” and “Do” below. Failure to do so may result in vessel trauma, guide wire damage, guide wire tip separation, or stent damage. If resistance is observed at any time, determine the cause under fluoroscopy and take remedial action as needed. Use the most suitable guide wire for the lesion being treated.
Do Not:
Do:
For the HI-TORQUE PROGRESS™ family only: The HI- TORQUE PROGRESS™ family of guide wires has distal ends of varying stiffness. Operate these guide wires carefully so as to not injure the blood vessel, observing the information in these instructions. The higher torque performance, stiffer distal ends, and / or higher advancement force may present a higher risk of perforation or injury than a guide wire with a more pliable distal end. Therefore, use the guide wire with the least stiff distal end that will treat the lesion, and use extreme care to minimize the risk of perforation or other damage to blood vessels.
PRECAUTIONS
Guide wires are delicate instruments and should be handled carefully. Prior to use and when possible during the procedure, inspect the guide wire carefully for bends, kinks, or other damage. Do not use damaged guide wires. Using a damaged guide wire may result in vessel damage and / or inaccurate torque response.
Confirm the compatibility of the guide wire diameter with the interventional device before actual use.
Free movement of the guide wire within the interventional device is an important feature of a steerable guide wire system, because it gives the user valuable tactile information. Test the system for any resistance prior to use. Adjust or replace the hemostatic valve with an adjustable valve if it is found to inhibit guide wire movement.
Never attach the torque device to the modified portion of the proximal end of the extendible guide wire; otherwise, guide wire damage may occur, preventing the ability to attach the DOC™ Guide Wire Extension.
Avoid abrasion of the hydrophilic coating. Do not withdraw or manipulate the hydrophilic-coated wire through a metal cannula or sharp-edged object.
ADVERSE EVENTS
Potential Adverse Events associated with use of this device may include the following, but not limited to: perforation, dissection, occlusion, myocardial infarction, embolism and infection.
MAT-2213673 v1.0
INTENDED USE
All HI-TORQUE™ Guide Wires are intended to facilitate the placement of balloon dilatation catheters during percutaneous transluminal coronary angioplasty (PTCA) and percutaneous transluminal angioplasty (PTA).
INDICATIONS
Refer to the device label for any additional product-specific indications that may apply.
CONTRAINDICATIONS
HI-TORQUE™ Guide Wires Wires are not intended for use in the cerebral vasculature. Refer to the device label for any additional product-specific contraindications that may apply.
WARNINGS
This device is designed and intended for ONE-TIME USE ONLY. Do not resterilize and / or reuse.
Carefully observe the instructions under “Do Not” and “Do” below. Failure to do so may result in vessel trauma, guide wire damage, guide wire tip separation, or stent damage. If resistance is observed at any time, determine the cause under fluoroscopy and take remedial action as needed. Use the most suitable guide wire for the lesion being treated.
Do Not:
Do:
PRECAUTIONS
Guide wires are delicate instruments and should be handled carefully. Prior to use and when possible during the procedure, inspect the guide wire carefully for bends, kinks, or other damage. Do not use damaged guide wires. Using a damaged guide wire may result in vessel damage and / or inaccurate torque response.
Confirm the compatibility of the guide wire diameter with the interventional device before actual use.
Free movement of the guide wire within the interventional device is an important feature of a steerable guide wire system, because it gives the user valuable tactile information. Test the system for any resistance prior to use. Adjust or replace the hemostatic valve with an adjustable valve if it is found to inhibit guide wire movement.
Never attach the torque device to the modified portion of the proximal end of the extendable guide wire; otherwise, guide wire damage may occur, preventing the ability to attach the DOC™ Guide Wire Extension.
HI-TORQUE™ Guide Wires with Hydrophilic Coating: Avoid abrasion of the hydrophilic coating.
Do not withdraw or manipulate the hydrophilic-coated wire through a metal cannula or sharp-edged object.
ADVERSE EVENTS
Potential Adverse Events associated with use of this device may include the following but not limited to perforation, dissection, occlusion, myocardial infarction, embolism and infection.
MAT-2213672 v1.0
The Teleport‡ microcatheters are indicated for:
The use of the Teleport‡ microcatheters are contraindicated:
When using this type of device, the following warnings should be observed:
Adverse events due to the use of this product include, but are not limited to, the following:
CAUTION: This product is intended for use by or under the direction of a physician. Prior to use, reference the Instructions for Use, inside the product carton (when available), at manuals.eifu.abbott or at eifu.orbusneich.com for more detailed information on Indications, Contraindications, Warnings, Precautions and Adverse Events. This material is intended for use with healthcare professionals only.
MAT-2400961 v2.0
INDICATIONS
Applies to OPTIS™ Imaging Systems and Software
The OPTIS™ Software and AptiVue™ E Series Software are intended to be used only with compatible OPTIS™ Imaging Systems.
The OPTIS™ Imaging Systems with a compatible Dragonfly™ Imaging Catheter are intended for the imaging of coronary arteries and is indicated in patients who are candidates for transluminal interventional procedures. The compatible Dragonfly™ Imaging Catheters are intended for use in vessels 2.0 to 3.5 mm in diameter. The compatible Dragonfly™ Imaging Catheters are not intended for use in the left main coronary artery or in a target vessel which has undergone a previous bypass procedure.
The OPTIS™ Imaging Systems are intended for use in the catheterization and related cardiovascular specialty laboratories and will further compute and display various physiological parameters based on the output from one or more electrodes, transducers, or measuring devices. The physician may use the acquired physiological parameters, along with knowledge of patient history, medical expertise and clinical judgment to determine if therapeutic intervention is indicated.
Applies to OPTIS™ Next Imaging Systems and Software
The Ultreon™ 1.0 Software and Ultreon™ 2.0 Software are intended to be used only with compatible OPTIS™ Next Imaging Systems.
The OPTIS™ Next Imaging System with a compatible Dragonfly™ OPTIS™ Imaging Catheter or Dragonfly OpStar™ Imaging Catheter is intended for the imaging of coronary arteries and is indicated in patients who are candidates for transluminal interventional procedures. The Dragonfly™ OPTIS™ Imaging Catheter or Dragonfly OpStar™ Imaging Catheter is intended for use in vessels 2.0 to 3.5 mm in diameter. The Dragonfly™ OPTIS™ Imaging Catheter or Dragonfly OpStar™ Imaging Catheter is not intended for use in the left main coronary artery or in a target vessel which has undergone a previous bypass procedure.
The OPTIS™ Next Imaging Systems are intended for use in the catheterization and related cardiovascular specialty laboratories and will further compute and display various physiological parameters based on the output from one or more electrodes, transducers, or measuring devices. The physician may use the acquired physiological parameters, along with knowledge of patient history, medical expertise, and clinical judgment to determine if therapeutic intervention is indicated.
Applies to both OPTIS™ and OPTIS™ Next Imaging Systems and Software
The Dragonfly™ OPTIS™ or Dragonfly™ OpStar™ Imaging Catheters are intended for use in vessels 2.0 to 3.5 mm in diameter. The Dragonfly™ OPTIS™ or Dragonfly™ OpStar™ Imaging Catheters are not intended for use in the left main coronary artery or in a target vessel which has undergone a previous bypass procedure.
The OPTIS™ and OPTIS™ Next Imaging Systems are intended for use in the catheterization and related cardiovascular specialty laboratories and will further compute and display various physiological parameters based on the output from one or more electrodes, transducers, or measuring devices. The physician may use the acquired physiological parameters, along with knowledge of patient history, medical expertise, and clinical judgment to determine if therapeutic intervention is indicated.
CONTRAINDICATIONS
The OPTIS™ and OPTIS™ Next Integrated Systems and Mobile Systems with the usage of the OPTIS™ Software, AptiVue™ E Series Software, Ultreon™ 1.0 Software, and Ultreon™ 2.0 Software are contraindicated where introduction of any catheter would constitute a threat to patient safety. Contraindications include:
COMPLICATIONS
The following complications may occur as a consequence of intravascular imaging and catheterization procedure:
WARNINGS
PRECAUTIONS
MAT-2309288 v1.0
Indications: The Dragonfly OpStar™ Imaging Catheter with the OCT Imaging System is intended for the imaging of coronary arteries and is indicated in patients who are candidates for transluminal interventional procedures. The Dragonfly OpStar™ Imaging Catheter is intended for use in vessels 2.0 to 3.5 mm in diameter. The Dragonfly OpStar™ Imaging Catheter is not intended for use in the left main coronary artery or in a target vessel which has undergone a previous bypass procedure.
Contraindications: Use of the Dragonfly OpStar™ Imaging Catheter is contraindicated where introduction of any catheter would constitute a threat to patient safety. Contraindications include:
Warnings:
Precautions:
Complications:
The following complications may occur as a consequence of intravascular imaging and catheterization procedure:
MAT-2115499 v3.0
Indications: CoroFlow‡ is indicated to provide hemodynamic information for use in the diagnosis of patients with cardiovascular diseases.
CoroFlow‡ is intended for use in catheterization and related cardiovascular specialty laboratories to compute and display various physiological parameters based on the output from one or more measuring devices.
Contraindications: The system has no patient alarm functions. Do not use for cardiac/vital signs monitoring.
Warnings:
Precautions:
MAT-2007904 v3.0
Indications: The PressureWire™ X Guidewire is indicated to direct a catheter through a blood vessel and to measure physiological parameters in the heart and in the coronary and peripheral blood vessels. Physiological parameters include blood pressure. The PressureWire™ X Guidewire can also measure blood temperature.
Contraindications: This guidewire is contraindicated for use in the cerebral vasculature.
Warnings:
Precautions:
Potential Adverse Events: Potential complications which may be encountered during all catheterization procedures include, but are not limited to: vessel dissection or occlusion, perforation, embolus, spasm, local and/or systemic infection, pneumothorax, congestive heart failure, myocardial infarction, hypotension, chest pain, renal insufficiency, serious arrhythmias, or death.
In addition, this device has a coating containing Polyethylene Glycol (PEG); potential allergic reactions (anaphylaxis) may occur during the interventional procedure if the patient is allergic to PEG.
MAT-2103599 v2.0
The NC TREK NEO™ Coronary Dilatation Catheters are indicated for:
a) balloon dilatation of the stenotic portion of a coronary artery or bypass graft stenosis, for the purpose of improving myocardial perfusion
b) balloon dilatation of a coronary artery occlusion, for the purpose of restoring coronary flow in patients with ST-segment elevation myocardial infarction
c) balloon dilatation of a stent after implantation (balloon models 2.00 mm – 5.00 mm only)
The NC TREK NEO™ Coronary Dilatation Catheter is contraindicated for treatment of the unprotected left main coronary artery and for coronary artery spasm in the absence of a significant stenosis
This device is intended for one time use only. DO NOT resterilize and / or reuse it, as this can compromise device performance and increase the risk of cross contamination due to inappropriate reprocessing.
Note the “Use by” date specified on the package.
The outside diameter (OD) of the distal 38 cm of the device, including the distal shaft, tip, and the balloon are coated with HYDROCOAT™ Hydrophilic Coating. Refer to PREPARATIONS FOR USE section of these instructions for further information on how to prepare and use this device to ensure it performs as intended. Failure to abide by the warnings in this labeling might result in damage to the device coating, which may necessitate intervention or result in serious adverse events.
Percutaneous transluminal coronary angioplasty (PTCA) should only be performed at centers where emergency coronary artery bypass graft surgery is available.
PTCA in patients who are not acceptable candidates for coronary artery bypass graft surgery requires careful consideration, including possible hemodynamic support during PTCA, as treatment of this patient population carries special risk.
Persons with known history of allergies to any of the components of this device listed below may suffer an allergic reaction to this coronary dilatation catheter. Prior to its use on the patient, the patient should be counseled on the materials contained in the device, and a thorough history of allergies must be discussed. This device contains: polyethylene oxide coating, polyamide, polyether block amide (PEBAX), polyethylene and stainless steel.
Use only the appropriate balloon inflation media. Do not use air or any gaseous medium to inflate the balloon. If gaseous medium is used and balloon rupture occurs there is a potential of causing air embolism and / or vessel injury. Balloon pressure should not exceed the rated burst pressure (RBP). Use of a pressure-monitoring device is recommended to prevent over pressurization.
To reduce the potential for vessel damage, the inflated diameter of the balloon should approximate the diameter of the normal or undiseased vessel segment, just proximal and distal to the stenosis.
Do not use or attempt to straighten a catheter if the shaft has become bent or kinked; this may result in the shaft breaking. Instead, prepare a new catheter.
Treatment of moderately or heavily calcified lesions is considered to be moderate risk, with increase in the risk of acute closure, vessel trauma, balloon burst, balloon entrapment, and associated complications. If resistance is felt, determine the cause before proceeding.
Continuing to advance or retract the catheter while under resistance may result in damage to the vessels and / or damage / separation of the catheter.
When the catheter is exposed to the vascular system, it should be manipulated while under high quality fluoroscopic observation. Do not advance or retract the catheter unless the balloon is fully deflated under vacuum. If resistance is met during manipulation, determine the cause of the resistance before proceeding.
Do not torque the catheter more than one (1) full turn.
In the event of catheter damage / separation, retrieval methods (use of additional wires, snares, and / or forceps) may result in additional trauma to the coronary vasculature and / or the vascular access site. Complications may include bleeding, hematoma, or pseudoaneurysm.
To confirm sterility has been maintained, ensure that the package sterile barrier has not been opened or damaged prior to use. Inspect all product and ensure that the device is not damaged. Care must be taken to properly size the balloon prior to use.
During the procedure, appropriate anticoagulant and coronary vasodilator therapy must be provided to the patient as needed. Anticoagulant therapy should be continued for a period of time as determined by the physician after the procedure.
If the surface of the coronary dilatation catheter becomes dry, wet with heparinized normal saline to reactivate the coating.
Do not reinsert the coronary dilatation catheter into the coil dispenser after procedural use
The safety and effectiveness of these devices have not been established, or is unknown, in vascular regions other than those specifically indicated:
Possible adverse effects include, but are not limited to, the following:
MAT-2206959 v3.0
CAREFULLY READ ALL INSTRUCTIONS PRIOR TO USE. OBSERVE ALL WARNINGS AND PRECAUTIONS NOTED THROUGHOUT THESE INSTRUCTIONS. FAILURE TO DO SO MAY RESULT IN COMPLICATIONS.
Applies to TREK™ RX & OTW 2.25 mm – 5.00 mm sizes only:
The TREK™ RX & OTW Coronary Dilatation Catheters are indicated for:
Applies to MINI TREK™ RX and MINI TREK™ II OTW
1.50 mm – 2.00 mm sizes only:
The TREK™ RX & OTW Coronary Dilatation Catheters are indicated for:
Applies to MINI TREK™ RX and MINI TREK™ II OTW 1.20 mm sizes only:
The MINI TREK™ RX and MINI TREK™ II OTW 1.20mm Coronary Dilatation Catheters are indicated for:
Note (applies to 2.00 mm to 5.00 mm only): Post-deployment stent expansion testing was performed on the bench with the MULTI-LINK VISION™ and MULTI-LINK ULTRA™ stents. All stents should be deployed in accordance with the manufacturer’s indications and instructions for use.
The TREK™ RX & OTW, MINI TREK™ RX and MINI TREK™ II OTW Coronary Dilatation Catheters are not intended to be used to treat patients with:
This device is intended for one time use only. DO NOT resterilize and / or reuse it, as this can compromise device performance and increase the risk of cross contamination due to inappropriate reprocessing.
Percutaneous transluminal coronary angioplasty (PTCA) should only be performed at hospitals where emergency coronary artery bypass graft surgery can bequickly performed in the event of a potentially injurious or life-threatening complication.
PTCA in patients who are not acceptable candidates for coronary artery bypass graft surgery requires careful consideration, including possible hemodynamic support during PTCA, as treatment of this patient population carries special risk.
Use only the recommended balloon inflation medium. Never use air or any gaseous medium to inflate the balloon.
Balloon pressure should not exceed the rated burst pressure (RBP). The RBP is based on results of in vitro testing. At least 99.9% of the balloons (with a 95% confidence) will not burst at or below their RBP. Use of a pressure-monitoring device is recommended to prevent overpressurization.
To reduce the potential for vessel damage, the inflated diameter of the balloon should approximate the diameter of the vessel just proximal and distal to the stenosis.
When the catheter is exposed to the vascular system, it should be manipulated while under high quality fluoroscopic observation. Do not advance or retract the catheter unless the balloon is fully deflated under vacuum. If resistance is met during manipulation, determine the cause of the resistance before proceeding.
Do not use, or attempt to straighten, a catheter if the shaft has become bent or kinked; this may result in the shaft breaking. Instead, prepare a new catheter.
Do not torque the catheter more than one (1) full turn.
Treatment of moderately or heavily calcified lesions is considered to be moderate risk, with an expected success rate of 60 – 85% and increases the risk of acute closure, vessel trauma, balloon burst, balloon entrapment, and associated complications. If resistance is felt, determine the cause before proceeding. Continuing to advance or retract the catheter while under resistance may result in damage to the vessels and / or damage / separation of the catheter.
In the event of catheter damage / separation, recovery of any portion should be performed based on physician determination of individual patient condition and appropriate retrieval protocol.
Note the “Use by” date specified on the package.
Inspect all product prior to use. Do not use if the package is open or damaged.
This device should be used only by physicians trained in angiography and PTCA, and / or percutaneous transluminal angioplasty (PTA).
Prior to angioplasty, the dilatation catheter should be examined to verify functionality and ensure that its size is suitable for the specific procedure for which it is to be used.
During the procedure, appropriate anticoagulant and coronary vasodilator therapy must be provided to the patient as needed. Anticoagulant therapy should be continued for a period of time to be determined by the physician after the procedure.
If the surface of the TREK™ RX & OTW, MINI TREK™ RX or MINI TREK™ II OTW Coronary Dilatation Catheter becomes dry, wetting with heparinized normal saline will reactivate the coating.
Do not reinsert the TREK™ RX & OTW, MINI TREK™ RX or MINI TREK™ II OTW Coronary Dilatation Catheter into the coil dispenser after procedural use.
The safety and effectiveness of this PTCA balloon catheter for the treatment of in-stent restenosis (ISR) have not been established.
Applies to TREK™ RX and MINI TREK™ RX only (APPLIES TO ALL SIZES), in addition to above:
The design and construction of these catheters do not provide the user with distal pressure monitoring capability.
Applies to TREK™ RX 4.50mm and5.00mm sizes only, in addition to above:
With 4.5 mm and 5.0 mm balloon dilatation catheters, some increased resistance may be noted upon insertion or withdrawal into or out of the guiding catheter. Choosing a larger guiding catheter size may minimize this.
Applies to TREK™ OTW and MINI TREK™ II OTW (APPLIES TO ALL SIZES), in addition to above:
Bench testing was conducted with 0.014” (.36mm) constant diameter guide wires to establish guide wire compatibility. If another type of guide wire is selected with a different dimensional profile, the compatibility (e.g., wire resistance) should be considered prior to use.
Possible adverse effects include, but are not limited to, the following:
MAT-2109405 v2.0
Indications
Applies to XIENCE Skypoint™ Stent Systems:
Applies to XIENCE Sierra™ and XIENCE Alpine™ Stent Systems:
Contraindications
The XIENCE Skypoint™, XIENCE Sierra™ and XIENCE Alpine™ Stent Systems are contraindicated for use in:
Warnings
Failure to abide by the warnings in this labeling might result in damage to the device coating, which may necessitate intervention or result in serious adverse events.
Precautions
Potential Adverse Events
Adverse events that may be associated with PCI treatment procedures and the use of a stent in native coronary arteries include, but are not limited to, the following:
The risks described below include the anticipated adverse events relevant for the cardiac population referenced in the contraindications, warnings and precaution sections of the everolimus labels / SmPCs and / or observed at incidences ≥ 10% in clinical trials with oral everolimus for different indications. Please refer to the drug SmPCs and labels for more detailed information and less frequent adverse events.
There may be other potential adverse events that are unforeseen at this time.
MAT-2100879 v7.0
Indications:
The Perclose™ ProStyle™ Suture-Mediated Closure and Repair System is indicated for the percutaneous delivery of suture for closing the common femoral artery and vein access sites of patients who have undergone diagnostic or interventional catheterization procedures.
The Perclose™ ProStyle™ SMCR System is indicated for closing the common femoral vein in single or multiple access sites per limb.
The Perclose™ ProStyle™ SMCR System is used without or, if required, with adjunctive manual compression.
For access sites in the common femoral artery using 5F to 21F sheaths. For arterial sheath sizes greater than 8F, at least two devices and the pre-close technique are required.
For access sites in the common femoral vein using 5F to 24F sheaths. For venous sheath sizes greater than 14F, at least two devices and the pre-close technique are required.
Caution:
Federal law restricts this medical device to sale by or on the order of a physician (or allied healthcare professionals, authorized by, or under the direction of, such physicians) who is trained in diagnostic and / or interventional catheterization procedures and who has been trained by an authorized representative of Abbott.
Prior to use, the operator must review the Instructions for Use and be familiar with the deployment techniques associated with the use of this device.
During closure of access sites using a procedural sheath greater than 8F, it is recommended that a vascular surgeon or a surgeon with vascular training be available in case surgical conversion to control bleeding and to repair the vessel is needed.
Contraindications:
There are no known contraindications to the use of this device.
Warnings:
Do not use the Perclose™ ProStyle™ SMCR System if the packaging or sterile barrier has been previously opened or damaged or if the components appear to be damaged or defective.
DO NOT RESTERILIZE OR REUSE. The Perclose™ ProStyle™ SMCR System is intended for single use only.
Do not use the Perclose™ ProStyle™ SMCR System if the sterile field has been broken where bacterial contamination of the sheath or surrounding tissues may have occurred, since such a broken sterile field may result in infection.
Do not use the Perclose™ ProStyle™ SMCR System if the puncture site is located above the most inferior border of the inferior epigastric artery (IEA) and / or above the inguinal ligament based upon bony landmarks, since such a puncture site may result in a retroperitoneal hematoma. Perform a femoral angiogram to verify the location of the puncture site. Note: This may require both a right anterior oblique (RAO) and left anterior oblique (LAO) angiogram to adequately visualize where the sheath enters the femoral vessel.
Do not use the Perclose™ ProStyle™ SMCR System in arterial or venous access if the puncture is through the posterior wall or if there are multiple punctures in the same access site, since such punctures may result in a hematoma or retroperitoneal bleed.
Do not use the Perclose™ ProStyle™ SMCR System if the puncture site is located in the superficial femoral artery or the profunda femoris artery, or the bifurcation of these vessels, since such puncture sites may result in a pseudoaneurysm, intimal dissection, or an acute vessel closure (thrombosis of small artery lumen). Perform a femoral angiogram to verify the location of the puncture site. Note: This may require both a right anterior oblique (RAO) and left anterior oblique (LAO) angiogram to adequately visualize where the sheath enters the femoral vessel.
Precautions:
Potential Adverse Events:
Potential adverse events associated with use of vessel closure devices may include, but are not limited to, the following:
MAT-2100368 v4.0
The StarClose SE™ Vascular Closure System is indicated for the percutaneous closure of common femoral artery access sites while reducing times to hemostasis, ambulation, and dischargeability in patients who have undergone diagnostic endovascular catheterization procedures utilizing a 5F or 6F procedural sheath.
The StarClose SE™ Vascular Closure System is indicated for use to allow patients who have undergone diagnostic endovascular catheterization procedures to ambulate and be eligible for discharge as soon as possible after device placement.
The StarClose SE™ Vascular Closure System is indicated for the percutaneous closure of common femoral artery access sites while reducing times to hemostasis and ambulation in patients who have undergone interventional endovascular catheterization procedures utilizing a 5F or 6F procedural sheath.
Federal law restricts this device to sale by or on the order of a physician (or allied healthcare professionals, authorized by, or under the direction of, such physicians) who is trained in diagnostic and therapeutic catheterization procedures and who has been trained by an authorized representative of Abbott Vascular.
Prior to use, the operators must review the Instructions for Use and be familiar with the deployment techniques associated with the use of this device.
The StarClose SE™ Vascular Closure System is contraindicated for use in patients with known hypersensitivity to nickel-titanium.
Do not use the StarClose SE™ Vascular Closure System if the packaging or sterile barrier has been previously opened or damaged or if the components appear to be damaged or defective.
DO NOT RESTERILIZE OR REUSE. The StarClose SE™ Vascular Closure System and accessories are intended for single use only.
Do not use the StarClose SE™ Vascular Closure System if the sterile field has been broken where bacterial contamination of the sheath or surrounding tissues may have occurred, since such a broken sterile field may result in infection.
Do not use the StarClose SE™ Vascular Closure System if the puncture site is located above the most inferior border of the inferior epigastric artery (IEA) and / or above the inguinal ligament based upon bony landmarks, since such a puncture site may result in a retroperitoneal hematoma. Perform a femoral angiogram to verify the location of the puncture site.
Do not use the StarClose SE™ Vascular Closure System if the puncture is through the posterior wall or if there are multiple punctures, since such punctures may result in a retroperitoneal hematoma.
Do not use the StarClose SE™ Vascular Closure System if the puncture site is located in the superficial femoral artery or the profunda femoris artery, since such puncture sites may result in a pseudoaneurysm, intimal dissection, or an acute vessel closure (thrombosis of small artery lumen). Perform a femoral angiogram to verify the location of the puncture site.
The StarClose Clip has been shown to be MR Conditional immediately following implantation. A patient with this implant can be scanned safely immediately after clip placement under the following conditions:
In non-clinical testing, the StarClose Clip produced a temperature rise of 0.5°C at maximum MR system-reported whole-body-averaged specific absorption rate (SAR) of 3 W/kg for 15 minutes of MR scanning in a 3 Tesla MR system using a transmit/receive body coil.
The MR image quality may be compromised if the area of interest is in the exact same area or relatively close to the position of the StarClose Clip. Therefore, optimization of MR imaging parameters to compensate for the presence of this implant may be necessary.
Potential adverse events that could be associated with the use of this device include:
MAT-2114590 v3.0
Including the Orbital Atherectomy Device (OAD) with GlideAssist™, Saline Pump, ViperWire Advance™ Coronary Guide Wire, and ViperWire Advance™ with Flex Tip Coronary Guide Wire
INDICATIONS
The Diamondback 360™ Coronary Orbital Atherectomy System (OAS) is a percutaneous orbital atherectomy system indicated to facilitate stent delivery in patients with coronary artery disease (CAD) who are acceptable candidates for PTCA or stenting due to de novo, severely calcified coronary artery lesions.
CONTRAINDICATIONS
Use of the OAS is contraindicated for use in the following situations:
WARNINGS
PRECAUTIONS
POTENTIAL ADVERSE EVENTS
Potential adverse events that may occur and/or require intervention include, but are not limited to:
Diamondback 360™ and Diamondback 360 Precision™ Coronary Orbital Atherectomy System are manufactured and distributed by Cardiovascular Systems, Inc. (CSI). CSI is a subsidiary of the Abbott Group of Companies.
MAT-2303956 v1.0
INDICATIONS
The Scoreflex‡ NC Scoring PTCA Catheter is indicated for: Balloon dilatation of a de novo stenotic portion of a coronary artery and in-stent restenosis in coronary arteries in patients evidencing coronary ischemia for the purpose of improving myocardial perfusion.
CONTRAINDICATIONS
The use of the Scoreflex‡ NC Scoring PTCA Catheter is contraindicated in the following patient types:
WARNINGS
When using this type of device, the following warnings should be observed:
PRECAUTIONS
ADVERSE EVENTS
Adverse events that may be associated with the use of this product include, but are not limited to, the following:
CAUTION: This product is intended for use by or under the direction of a physician. Prior to use, reference the Instructions for Use, inside the product carton (when available), at manuals.eifu.abbott or at eifu.orbusneich.com for more detailed information on Indications, Contraindications, Warnings, Precautions and Adverse Events. This material is intended for use with healthcare professionals only.
Scoreflex‡ NC Scoring PTCA Catheter is manufactured by OrbusNeich Medical Group Holdings Limited or its affiliates and distributed by Cardiovascular Systems, Inc. (CSI). CSI is a subsidiary of the Abbott Group of Companies.
MAT-2303959 v2.0
INDICATIONS
The Sapphire‡ NC 24 Coronary Dilatation Catheter is indicated for:
CONTRAINDICATIONS
The use of Sapphire‡ NC 24 Coronary Dilatation Catheter is contraindicated in the following patient types:
WARNINGS
When using this type of device, the following warnings should be observed:
PRECAUTIONS
POTENTIAL COMPLICATIONS AND ADVERSE EVENTS
Potential complications and adverse effects due to the use of this product include, but are not limited to, the following:
CAUTION: This product is intended for use by or under the direction of a physician. Prior to use, reference the Instructions for Use, inside the product carton (when available), at manuals.eifu.abbott or at eifu.orbusneich.com for more detailed information on Indications, Contraindications, Warnings, Precautions and Adverse Events. This material is intended for use with healthcare professionals only.
MAT-2400959 v2.0
The Sapphire‡ II PRO Dilatation Catheter (Ø1.0-1.25mm configurations) is indicated for:
The Sapphire‡ II PRO Dilatation Catheter (Ø1.5-4.0mm configurations) is indicated for:
The Sapphire‡ II PRO Dilatation Catheter is also indicated for:
The use of the Sapphire‡ II PRO Dilatation Catheter is contraindicated:
When using this type of device, the following warnings should be observed:
PRECAUTIONS
ADVERSE EVENTS
Adverse effects due to the use of this product include, but are not limited to, the following:
CAUTION: This product is intended for use by or under the direction of a physician. Prior to use, reference the Instructions for Use, inside the product carton (when available), at manuals.eifu.abbott or at eifu.orbusneich.com for more detailed information on Indications, Contraindications, Warnings, Precautions and Adverse Events. This material is intended for use with healthcare professionals only.
MAT-2400936 v2.0
Stay Connected